Gauchersjukdom (uttalad "GO shay" -sjukdom) är ett genetiskt tillstånd med ett brett spektrum av kliniska symptom som påverkar flera organsystem i kroppen. I den vanligaste formen av Gaucher har människor mycket behandlingsbara symtom. I andra typer av Gauchers sjukdom är symptomen allvarliga och mycket svåra att behandla. Din läkare hjälper dig att lära dig vad du kan förvänta dig i din speciella situation.
Orsaker
Gauchers sjukdom är en genetisk sjukdom som orsakas av ett problem med en gen som kallas GBA. Denna gen är en del av ditt DNA, det genetiska materialet du arv från dina föräldrar.
GBA -genen är ansvarig för att göra ett enzym som kallas glukokrebrosidas. Hos människor med Gauchers sjukdom är detta enzym brist, eller det fungerar inte så bra som det borde. För att förstå vikten av detta enzym är det viktigt att veta om en del av cellen som kallas lysosomen. Lysosomer finns som komponenter inuti kroppens celler. De hjälper till att städa upp och kassera material som kroppen annars inte kan bryta ner. De spelar en viktig roll för att bryta ner material som kan ackumuleras i kroppen. Glukocerebrosidas är en av de enzymer som hjälper lysosomer att göra detta.
Normalt hjälper detta enzym till att återvinna ett fettämne i kroppen som kallas glukocerebroside. Men i Gauchers sjukdom fungerar inte glukokrebrosidas mycket bra.
Enzymet är kanske inte alls aktivt, eller det kan ha nedsatt aktivitet. På grund av detta börjar glukocerebrosidan att byggas upp i olika delar av kroppen. Detta leder till symptom på tillståndet.
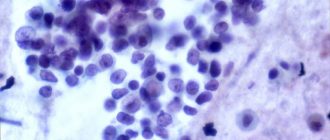
När vissa immunceller fylls med överskott av glukocerebroside kallas de "Gaucher-celler". Dessa Gaucher-celler kan massera ut normala celler och orsaka problem.
Uppbyggnaden av Gaucher-celler i benmärgen hindrar till exempel din kropp från att kunna producera de normala mängderna nya blodkroppar där. En uppbyggnad av glukocerebroside och Gaucher-celler är särskilt ett problem i mjälten, leveren, benet och hjärnan.
Problem med andra typer av enzymer i lysosomer kan leda till andra typer av störningar. Som en grupp kallas dessa lysosomala lagringssjukdomar.
Prevalens
Gauchersjukdom är ett sällsynt tillstånd. Det påverkar ungefär ett spädbarn född av 100.000. Men i vissa etniska grupper är Gauchers sjukdom vanligare, som i Ashkenazi-judarna. Till exempel har ungefär en av 450 barn av denna genetiska bakgrund Gaucher-sjukdom.
Gauchersjukdom är den vanligaste av lysosomala förvaringssjukdomar, som inkluderar andra tillstånd såsom Tay-Sachs sjukdom och Pompe-sjukdom.
Diagnos
En läkare kan först misstänka Gauchers sjukdom baserat på en persons symptom och medicinska tecken. Om en person är känd att ha Gaucher-sjukdom i sin familj, ökar misstanken om sjukdomen.
Människor med Gauchersjukdom har ofta ovanliga laboratoriefinster, såsom på benmärgsfärg. Dessa resultat kan vara till hjälp för att peka mot Gaucher.
Det finns en mängd andra laboratorie- och bildtesttest som din läkare kan använda för att utvärdera statusen för din Gaucher. Till exempel kan din läkare vilja ha en MR för att kontrollera för inre organförstoring.
För en sann diagnos behöver din läkare också ett blodprov eller en hudbiopsi. Detta prov används för att se hur bra glukokrebrosidas fungerar. Ett alternativ är ett genetiskt blod eller vävnadstest som används för att analysera GBA-genen.
Eftersom det är en sällsynt sjukdom, är de flesta läkare inte särskilt bekanta med Gaucher. Delvis på grund av detta tar diagnosen Gauchersjukdom ibland ett tag.
Detta är speciellt troligt om ingen annan i familjen redan är känd för att ha det.
Typer
Det finns tre huvudtyper Gauchersjukdom: typ 1, typ 2 och typ 3. Dessa typer skiljer sig något i deras symtom och i deras svårighetsgrad. Typ 1 är den mildaste formen av Gaucher. Det påverkar inte nervsystemet, till skillnad från typ 2 och typ 3 Gaucher sjukdom. Typ 2 Gauchersjukdom är den mest allvarliga typen.
En stor majoritet av personer med Gaucher-sjukdom har typ 1-sjukdom. Cirka 1 procent av personer med Gaucher anses ha typ 2-sjukdom. Omkring 5 procent av personer med Gaucher har typ 3-sjukdom.
När man överväger symtom på Gauchers sjukdom, är det viktigt att komma ihåg att människor upplever ett stort antal symptomsvårigheter. Symtom överlappar mellan de tre typerna.
Symptom på typ 1
Tecken och symtom på typ 1 Gauchersjukdom uppträder först i barndomen eller vuxen ålder. Benproblem kan innehålla:
Kronisk benvärk
- Plana biverkningar av benvärk
- Benfraktur
- Osteoporos
- Artrit
- Typ 1 Gaucher påverkar också några av de inre organen. Det kan orsaka utvidgning av mjälten och leveren (kallad hepatosplenomegali). Detta är vanligtvis smärtfritt men orsakar bukdistension och en känsla av fullhet.
Typ 1 Gaucher orsakar också något som kallas cytopeni. Detta innebär att personer med Gaucher-sjukdom har lägre nivåer än röda blodkroppar (orsakar anemi), vita blodkroppar och blodplättar. Människor med Gaucher kan också ha andra koagulations- och immunförsvar. Detta kan leda till symtom som:
Utmattning
- Lätt blödning eller blåmärken
- Näsblod
- Ökad risk för infektion
- Gauchers sjukdom kan också påverka lungorna, vilket leder till sådana problem som:
Interstitiell lungsjukdom
- Pulmonell hypertension
- Hosta ▲ Andnöd
- Dessutom kan typ 1 Gaucher orsaka:
- Ökad risk för gallstones
Dålig tillväxt och utveckling
- Psykologiska komplikationer, såsom nedsatt humör
- Hjärtekomplikationer (sällsynta). Njurkomplikationer (sällsynta). Några personer som har typ 1 Gaucher-sjukdom har mycket mild sjukdom och kanske inte märker några symtom. Dock kan kliniker upptäcka små abnormiteter med hjälp av laboratoriefynd och bildbehandlingstester.
- Symtom på typ 2 och 3
- Nästan alla samma system i kroppen som drabbas av typ 1-sjukdom kan också orsaka problem vid typ 2 och typ 3-sjukdom. Men typ 2 och 3 har också ytterligare neurologiska symptom. Dessa symptom är störst hos patienter med typ 2-sjukdom. Dessa barn dör vanligen före ålder 2. I en mycket sällsynt form av sjukdomen dör barn kort före eller kort efter födseln. Hos människor med typ 3 Gaucher är dessa problem inte lika svåra, och människor kan leva i 20-årsåldern, 30-talet eller längre.
- Neurologiska symtom som ses i typ 2 och typ 3-sjukdom, många inkluderar:
Ögonviktning (strabismus)
Problem med spårning av föremål eller skiftande blick
Sprickor
Muskelstyvhet
- Muskelsvaghet
- Problem med balans och samordnad rörelse
- Problem med tal och sväljning
- Problem med tal och sväljning
- Mental retardation
- Demens
- En delmängd av personer med typ 2 eller typ 3 Gaucher har också ytterligare symtom. Exempel är hudförändringar, problem med deras hornhinna och hjärtklaffförkalkning.
- Sekundära sjukdomar
- Gauchersjukdom ökar också risken för andra sjukdomar. Till exempel har personer med Gaucher en högre än genomsnittlig risk för Parkinsons sjukdom. Vissa cancers kan också vara vanligare hos personer med Gauchers sjukdom, inklusive:
Blodcancer
Multipel myelom
Levercancer
- Njurcancer
- Människor med Gaucher har också risk för några sekundära komplikationer, såsom mjältinfarkt (brist på blodflöde till mjälten, vilket orsakar vävnadsdöd och svår magsmärta).
- Behandling
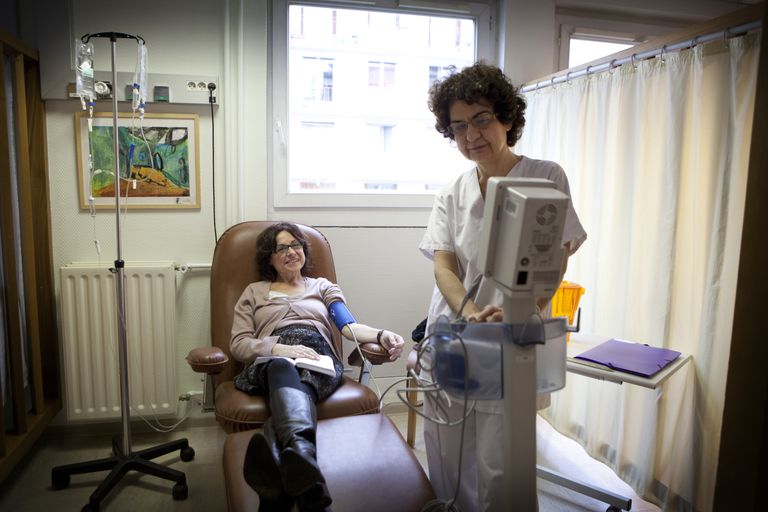
- Behandlingsstandarden för Gaucher-sjukdomen är enzymbytebehandling (ibland kallad ERT). Denna behandling revolutionerade behandlingen av Gaucher. I ERT mottager en person en konstgjord syntesform av glukokrebrosidas i form av en intravenös infusion. Olika former av ERT finns nu på marknaden kommersiellt, men alla tillhandahåller ersättningsenzym. Dessa är:
imigluceras (varumärke som Cerezyme)
velagluceras alfa (VPRIV)
taligluceras alfa (Elelyso)
Dessa behandlingar är mycket effektiva vid minskning av bensymptom, blodproblem och lever- och mjältförstoring. Men de fungerar inte mycket bra på att förbättra de neurologiska symptomen som ses i typ 2 och typ 3 Gaucher sjukdomar.
- ERT är mycket effektivt för att minska symtomen på typ 1 Gaucher och att minska några av symtomen på typ 3 Gaucher. Tyvärr, eftersom typ 2 Gaucher har så svåra neurologiska problem rekommenderas inte ERT för denna typ. Personer med typ 2 Gaucher får vanligtvis endast stödjande behandling.
- Ett annat nyare behandlingsalternativ för typ 1 Gaucher är substratreduceringsbehandling. Dessa läkemedel begränsar produktionen av ämnen som glukocerebrosidas bryter ner. Dessa är:
- miglustat (Zavesca)
eliglustat (Cerdelga)
Miglustat finns som ett alternativ för personer som inte kan ta ERT av någon anledning. Eliglustat är ett oral läkemedel som är ett alternativ för vissa personer med typ 1 Gaucher. Det är ett nyare läkemedel, men vissa bevis tyder på att det är lika effektivt som ERT-behandlingar.
Dessa behandlingar för Gaucher kan vara mycket dyra. De flesta kommer att behöva arbeta nära sitt försäkringsbolag för att se till att de får tillräcklig täckning av behandlingen.
- Människor med Gauchers sjukdom ska behandlas av en specialist med erfarenhet av tillståndet. Dessa människor behöver regelbunden uppföljning och övervakning för att se hur väl deras sjukdom svarar på behandlingen. Till exempel behöver personer med Gaucher ofta upprepade benskanningar för att se hur sjukdomen påverkar deras ben.
- Människor som inte kan ta emot ERT eller en nyare substratreduktionsterapi kan behöva ytterligare behandlingar för symtomen på Gaucher. Till exempel kan dessa människor behöva blodtransfusioner för svår blödning.
Genetik
Gauchersjukdom är ett autosomalt recessivt genetiskt tillstånd. Det betyder att en person med Gaucher-sjukdom får en kopia av en drabbad
GBA
-gen från varje förälder. En person som bara har en kopia av en drabbad
GBA
-gen (ärvd från en förälder) sägs vara en bärare av tillståndet. Dessa människor har tillräckligt fungerande glukocerebrosidas att de inte har symptom. Sådana människor vet ofta inte att de är sjukdomsbärare såvida inte någon i deras familj diagnostiseras med sjukdomen. Bärare har risk att överföra en drabbad kopia av genen till sina barn. Om du och din partner båda är bärare för Gauchers sjukdom, finns det 25 procent chans att ditt barn kommer att få sjukdomen. Det finns också en 50 procent chans att ditt barn inte kommer att få sjukdomen men kommer också att vara en bärare för tillståndet. Det finns en 25 procent chans att ditt barn inte kommer att få sjukdomen eller vara en bärare. Prenatal testning är tillgänglig i fall där barnet riskerar Gaucher. Prata med din läkare om du oroar dig för att du kan vara en bärare av Gaucher-sjukdom baserat på din familjhistoria. Om någon i din familj har Gaucher-sjukdom, kan du vara i fara. Genetiska tester kan användas för att analysera dina gener och se om du är en sjukdomsbärare. Ett ord från Verywell Det kan vara överväldigande att lära dig att du eller en älskad har Gaucher-sjukdom. Det finns mycket att lära sig om hantering av tillståndet, och du behöver inte göra allt på en gång. Lyckligtvis, eftersom tillgången på ERT, kan många människor med Gaucher-sjukdom leda relativt normala liv.